Resources for Reusing Tools and Scripts
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How to find other tools/solutions for awkward problems?
Objectives
tools objectives:
know good resources for finding solutions to common problems
Learning objectives
By the end of this episode, learners should be aware of where they can look for CWL recipes and more help for common, but awkward, tasks.
Pre-written tool descriptions
When you start a CWL workflow, it is recommended to check if there is already a CWL document available for the tools you want to use. Bio-cwl-tools is a library of CWL documents for biology/life-sciences related tools.
The CWL documents of the previous steps were already provided for you, however, you can also find them in this library. In this episode you will use the bio-cwl-tools library to add the last step to the workflow.
Adding new step in workflow
The last step of our workflow is counting the RNA-seq reads for which we will use the featureCounts tool.
Exercise
Find the
featureCountstool in the bio-cwl-tools library. Have a look at the CWL document. Which inputs does this tool need? And what are the outputs of this tool?Solution
The
featureCountsCWL document can be found at https://github.com/common-workflow-library/bio-cwl-tools/blob/release/subread/featureCounts.cwl ; it has 2 inputs:annotations(line 6) andmapped_reads, both files. These inputs can be found on lines 6 and 9. The output of this tool is a file calledfeaturecounts(line 21).
After finding the featureCounts tool, we need to download the tool to the directory in which our workflow is located.
To download the tool, we go to the featureCounts.cwl GitHub page and open the raw file.
The screenshot below shows where you can find this button.
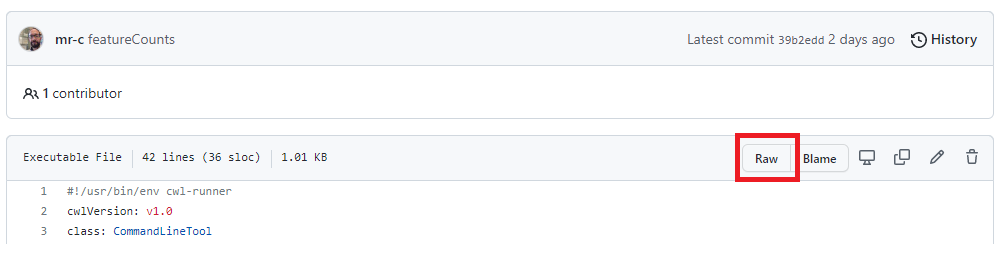
There are two approaches to download the CWL document.
- Copy the script to a new file in an editor, for example VSCode, and save the document in your directory.
- Use the
wgetcommand in the command line. You need to copy the URL of the raw file and use this command:wget [URL]
Exercise
Use either of the two approaches above to download the
featureCountstool and add thefeatureCountstool to the workflow. Similar to theSTARtool, this tool also needs more RAM than the default. To run the tool a minimum of 500 MiB of RAM is needed. Use arequirementsentry withResourceRequirementto allocate aramMinof 500. Use the inputs and output of the previous exercise to connect this step to previous steps.Solution
clwVersion: v1.2 class: Workflow inputs: rna_reads_human: File ref_genome: Directory annotations: File steps: quality_control: run: bio-cwl-tools/fastqc/fastqc_2.cwl in: reads_file: rna_reads_human out: [html_file] mapping_reads: requirements: ResourceRequirement: ramMin: 9000 run: bio-cwl-tools/STAR/STAR-Align.cwl in: RunThreadN: {default: 4} GenomeDir: ref_genome ForwardReads: rna_reads_human OutSAMtype: {default: BAM} SortedByCoordinate: {default: true} OutSAMunmapped: {default: Within} out: [alignment] index_alignment: run: bio-cwl-tools/samtools/samtools_index.cwl in: bam_sorted: mapping_reads/alignment out: [bam_sorted_indexed] count_reads: requirements: ResourceRequirement: ramMin: 500 run: bio-cwl-tools/subreads/featureCounts.cwl in: mapped_reads: index_alignment/bam_sorted_indexed annotations: annotations out: [featurecounts] outputs: qc_html: type: File outputSource: quality_control/html_file bam_sorted_indexed: type: File outputSource: index_alignment/bam_sorted_indexed featurecounts: type: File outputSource: count_reads/featurecounts
The workflow is complete and we only need to complete the YAML input file.
The last entry in the input file is the annotations file.
workflow_input.yml
rna_reads_human:
class: File
location: rnaseq/raw_fastq/Mov10_oe_1.subset.fq
format: http://edamontology.org/format_1930
ref_genome:
class: Directory
location: rnaseq/hg19-chr1-STAR-index
annotations:
class: File
location: rnaseq/reference_data/chr1-hg19_genes.gtf
You have finished the workflow and the input file and now you can run the whole workflow.
cwltool rna_seq_workflow.cwl workflow_input.yml
Key Points
First key point. Brief Answer to questions. (FIXME)